
If you run a U.S. peptide clinic, your adverse event log needs to do more than list side effects. It must support classification, escalation, reporting, follow-up, and record retention.
I’d boil the article down to this: you should log every event the same way, classify it at entry, move serious and unexpected cases on time, and keep the full case file for 10 years. In research settings, timing can be 7 or 15 calendar days under FDA IND rules. In postmarketing settings, serious and unexpected cases generally move within 15 calendar days. For compounded peptides, I’d also notify the compounding pharmacy within 24 hours when an event may point to a lot or product issue.
Here’s the plain-English version:
- Use one standard for every case: seriousness, severity, expectedness, and causality are not the same thing.
- Record the same core fields every time: patient, product, event details, assessment, and report dates.
- Keep source records tied to one case ID: chart notes, labs, discharge papers, follow-up calls, and updates.
- Build for the strictest rule that may apply: FDA, IRB, OHRP, or postmarketing safety rules.
- Match your workflow to fixed deadlines: document within 24 hours, review within 48 hours, and report when the case meets the trigger.
- Store the full record for 10 years: including raw data and related correspondence.
- Get ready for E2B(R3): FDA safety report formatting rules tighten in 2026.
I see the article as a compliance playbook, not just a documentation guide. The main point is simple: a clinic AE log should let someone reconstruct the case, decide if it is reportable, show what the clinic did, and prove that deadlines were met.
Submitting IND Safety Reports to FDA Adverse Event Reporting System (FAERS)- Nov. 1, 2019

sbb-itb-7164bd9
Regulatory Framework for Adverse Event Documentation
There isn’t one rulebook for peptide clinic AE logs. What you need to document depends on the setting: IND research, IRB/OHRP-covered research, or routine clinical care. In a peptide therapy clinic, the same event might need to be reported to a sponsor, an IRB, or only through an internal process based on how the peptide is being used. So the practical move is simple: build the log to meet the toughest rule that could apply.
FDA, IRB, and OHRP Requirements That Apply to AE Logs
When a peptide is given under an Investigational New Drug (IND) application, 21 CFR Part 312 sets the baseline. Under 21 CFR 312.62, investigators must keep complete case histories and promptly report adverse effects that are reasonably related to the drug to the sponsor. If the event is alarming, it must be escalated at once.
IRB and OHRP oversight adds a second layer. Under 21 CFR Part 56 and 45 CFR Part 46, investigators must report unanticipated problems involving risk to human subjects or others to the IRB. The FDA focuses on drug safety. IRB and OHRP focus on unanticipated risk to human subjects.
If more than one rule applies, the AE log should support the strictest reporting duty. Here’s the short version:
| Regulatory Body | Primary Regulation | Reporting Focus | Key Timeline |
|---|---|---|---|
| FDA (IND) | 21 CFR 312 | Serious and unexpected suspected adverse reactions | 7 or 15 calendar days |
| IRB | 21 CFR 56 | Unanticipated problems involving risk to subjects | Per IRB policy |
| OHRP | 45 CFR 46 | Unanticipated problems in federally funded research | Promptly |
| FDA (Marketed) | 21 CFR 310.305 | Serious, unexpected adverse drug experiences | 15 calendar days |
That setup only works if staff log events with the same terms and the same cutoffs. If one person marks an event as “serious” and another uses that label more loosely, reporting can go off the rails fast.
For marketed prescription drugs without an approved NDA, 21 CFR § 310.305 requires manufacturers and distributors to keep records of serious, unexpected adverse drug experiences for 10 years.
ICH Definitions Used in U.S. Research Workflows
Once the reporting path is clear, the next step is consistency. U.S. research programs use ICH E2A definitions so adverse events are classified the same way across cases. Each event should be classified by:
- Seriousness
- Expectedness
- Attribution
Seriousness covers outcomes like death, life-threatening conditions, hospitalization, disability, or congenital anomaly. Expectedness asks whether the event appears in the Investigator's Brochure. Attribution asks whether there is a reasonable possibility that the product caused the event. Those three calls shape where the report goes and how fast it needs to move. The key point: apply these definitions when staff enter the event, not later during review.
As of April 2026, IND sponsors must submit individual case safety reports using the ICH E2B(R3) electronic data standard. Starting October 1, 2026, all postmarketing ICSRs submitted through the FDA's ESG NextGen must also use E2B(R3). That’s why it makes sense to align internal AE log fields with E2B(R3) now. It cuts friction later when a case needs to be reported outside the clinic.
Standard Definitions and Minimum Data Fields for AE Logs
Once the terms are set, the log needs a minimum set of fields so each event is recorded the same way, every time. A compliant AE log depends on two things: using the right terms the same way and recording the right data fields for each case. If staff members apply definitions differently from one case to the next, the log stops being dependable. After classification is fixed, the next step is a format that treats every event the same way.
How to Classify Events: Seriousness, Severity, Expectedness, and Attribution
Do not treat severity and seriousness as the same thing. Severity is about intensity. Seriousness is about the regulatory outcome. An event is serious only if it results in death, a life-threatening experience, inpatient hospitalization or prolongation of hospitalization, persistent or significant disability or incapacity, a congenital anomaly or birth defect, or an important medical event that needs intervention to prevent one of those outcomes.
A symptom can be severe without being serious. That one difference changes the reporting path.
Expectedness and attribution finish the classification. Unexpected means the event is missing from current labeling or is more severe or more specific than what the label describes. Attribution asks whether there is a reasonable possibility of causation. These three classifications should be applied at the time of entry, not saved for a later review.
Core Fields Every Adverse Event Log Should Include
Each entry in a compliant AE log should include the same core data, whether the event later turns out to be reportable or not. For a valid FDA submission, the required elements are an identifiable patient, a suspect drug or biological product, the adverse event or report of death, and an identifiable reporter. At the clinic level, logs often include more detail to support internal review and audit readiness.
| Category | Minimum Required Fields |
|---|---|
| Patient Info | Unique patient ID code, age at time of event (or date of birth), gender, weight |
| Event Details | Onset date, resolution date, outcome (recovered/resolved, recovering/resolving, not recovered/not resolved, recovered with sequelae, or fatal), concise medical narrative, specific AE terms, relevant lab or test results |
| Product Info | Peptide name, dose, frequency, route of administration, therapy start/stop dates, lot number, expiration date, source pharmacy/manufacturer identifier or N/A |
| Clinical Assessment | Seriousness criteria, severity, expectedness vs. label, attribution, dechallenge/rechallenge results |
| Administrative | Date clinic received report, reporter contact info, clinician or PI review signature/date |
For compounded products, the lot number and expiration date matter even more. If an AE involves a compounded peptide, the compounding pharmacy should be notified within 24 hours so it can investigate possible batch-wide quality or contamination issues.
Use specific clinical language in the narrative field. For example:
"Sharp abdominal pain 30 minutes after injection", not "felt bad".
That kind of wording makes the record far more useful during review. For clinics needing standardized documentation, the Peptide Prescriber Starter Pack includes regulatory quick references and implementation tools.
Source Documentation and Audit Trail Standards
An AE log is only as solid as the records behind it. Every logged event should be backed by the clinical chart, hospital discharge summaries, lab data, or other records that show what happened and when.
Use one case ID and carry it through every update. If new details come in after the first entry, such as a lab result, follow-up call, or resolution date, add that information as a dated update rather than replacing the first record. If new information cannot be obtained, document each follow-up attempt.
The goal is simple: the log should let someone reconstruct the case and escalate it at any point in the review process.
Electronic and paper systems are both acceptable if they preserve completeness, accuracy, and traceable updates that authorized FDA personnel can review. That audit trail is what makes later escalation, review, and inspection defensible.
Reporting Timelines, Escalation Triggers, and Record Retention
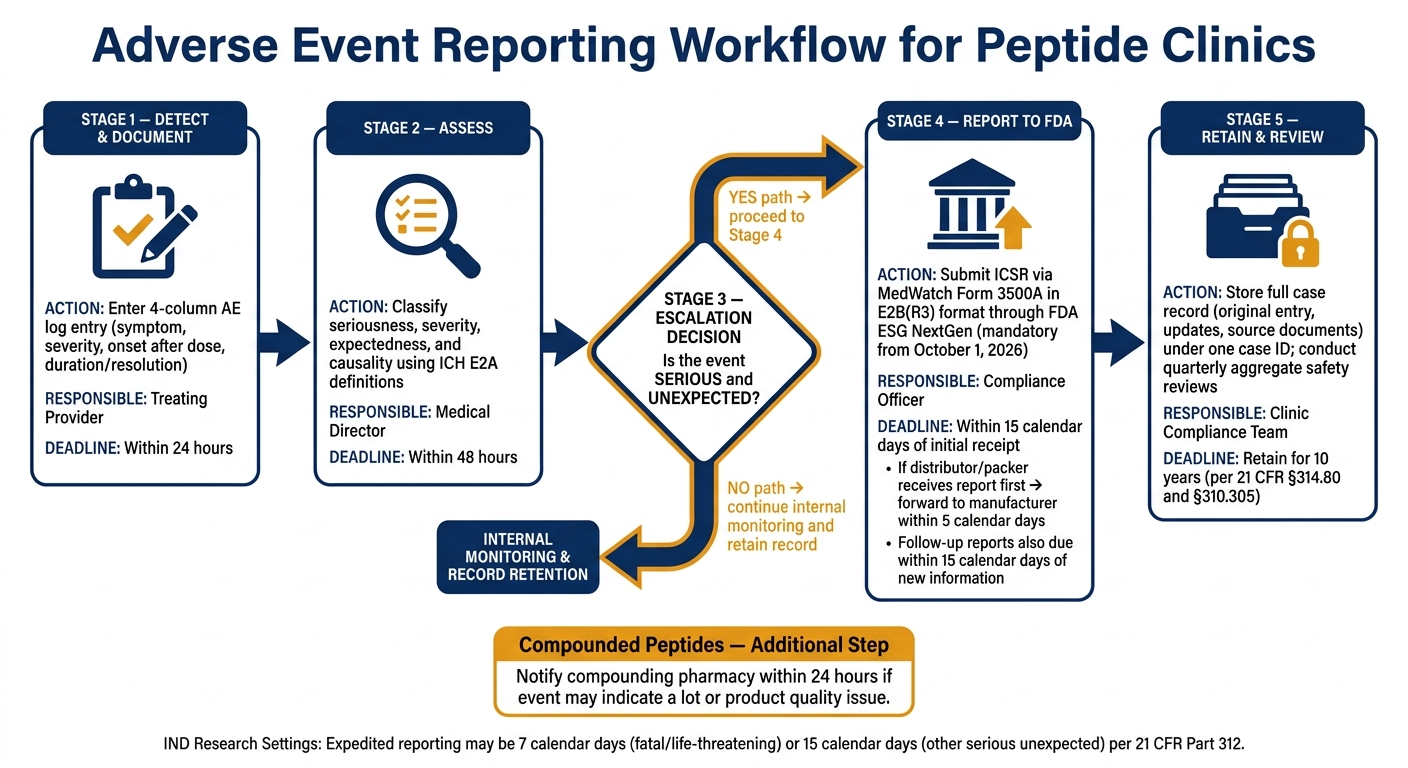
Adverse Event Reporting Workflow for Peptide Clinics
After entry and classification, the next step is reporting, escalation, and retention. Once the AE is logged and classified, the team needs to decide if it calls for expedited reporting.
Which Logged Events Require Rapid Reporting
Not every logged event needs to be sent to the FDA. For marketed drugs, expedited reporting is triggered when an event is both serious and unexpected. When that threshold is met, the event must be reported through MedWatch using Form 3500A for mandatory reports within 15 calendar days of initial receipt.
Document the pharmacy notification in the case file. If a distributor or packer receives the report first, it must forward the report to the manufacturer within 5 calendar days of receipt.
How to Build a Reporting Workflow From Log Entry to Submission
A clear internal sequence keeps events moving from detection to submission without delay. The table below shows each compliance-critical step, who owns it, when it must happen, and what needs to be documented.
| Internal Step | Responsible Party | Timeline | Documentation Requirement |
|---|---|---|---|
| Documentation | Treating provider | Within 24 hours | 4-column AE log entry |
| Assessment | Medical director | Within 48 hours | Determination of seriousness and causality |
| FDA Submission | Compliance officer | Within 15 calendar days | ICSR in E2B(R3) format |
If new information comes in after an initial 15-day alert report, the follow-up report must be sent to the FDA within 15 calendar days of receipt.
Starting October 1, 2026, all postmarketing individual case safety reports submitted through the FDA's Electronic Submissions Gateway Next Generation (ESG NextGen) must use E2B(R3) data standards.
After submission, keep the full case record under the same case ID.
Retention, Inspection Readiness, and Aggregate Safety Review
Because the log needs to support inspection and follow-up, retention is a compliance task, not just file storage. Federal regulations under 21 CFR § 314.80 and § 310.305 require that all adverse drug experience records, including raw data and related correspondence, be kept for 10 years. Keep the original entry, later updates, and source documents together under one case ID.
Store records securely and limit access to approved personnel. Electronic files should be backed up, and paper files should stay in locked storage.
Use quarterly reviews to drive protocol updates.
Implementing a Research-Grade AE Log in a Peptide Therapy Clinic
Minimum Clinic Standard for Peptide AE Logging
Once you know the minimum regulatory bar, the next step is turning it into a clinic process staff can use every single time. The goal isn't just internal note-taking. It's documentation that holds up under review. A simple five-step workflow works well: detect the event, document it within 24 hours, assess it within 48 hours, report serious events within 15 calendar days, and keep following the case until it resolves.
After that, lock down what gets entered for every case. Use a four-field template that includes:
- symptom
- severity (mild, moderate, severe)
- onset after dose
- duration/resolution
For each event, also record the peptide name, lot number, and expiration date from the vial label. That makes it much easier to tie a case back to the product source fast. If the product was compounded, notify the compounding pharmacy within 24 hours.
Quarterly review matters too. Look at aggregate adverse event data every quarter to spot repeat patterns tied to lots, doses, or clinic protocols, then update the protocol based on what you find. If the log never changes how the clinic works, it's just paperwork. That quarterly review should feed straight back into the template and EHR workflow.
Using Templates, EHR Workflows, and compliance resources
To cut down on missed fields, build the AE form directly into the EHR and use the same case ID for every update. Structured fields help prevent gaps and make escalation simpler. Each entry should link back to the chart, labs, photos, and product label under one case ID that stays with the case through all follow-up.
PeptidePrescriber offers licensed prescribers evidence-based clinical resources, regulatory references, and clinical tools supporting peptide therapy documentation workflows.
A defensible AE log comes down to doing the same thing every time: use standardized definitions when the event is entered, capture the required fields for each case, connect every entry to its source records, and escalate serious and unexpected events to the FDA within 15 calendar days.
FAQs
What makes an adverse event reportable?
An adverse event is any unexpected experience or side effect that happens after treatment, whether or not it’s believed to be linked to the product. That can include cases involving overdose, abuse, withdrawal, professional use, or a product that didn’t work as expected.
For FDA reporting, the event must be serious and unexpected. A serious event includes death, a life-threatening condition, hospitalization, disability, birth defects, or medical treatment needed to stop one of those outcomes from happening.
How should clinics handle follow-up updates?
Clinics should keep following up with patients after an adverse event to check on recovery and safety. The treating provider should review the patient’s condition and record the outcome in both the medical record and the adverse event file.
For serious, unexpected adverse events reported to the FDA, providers must investigate without delay and send follow-up reports within 15 calendar days of getting new information or when the FDA asks for it. If more information can’t be obtained, document the steps taken to try.
What records should be kept for each case?
For each adverse event, clinics should keep full records in the patient’s medical chart and in a separate adverse event file.
Write down:
- what happened
- when it happened
- the symptoms that were reported
- the treatment that was given
- how the patient responded
- any changes to the future treatment plan
It also helps to document follow-up efforts, including attempts to get more details that didn't lead anywhere.
For serious, unexpected events, the record should include the identifiable patient, identifiable reporter, the suspect drug, and the adverse event.